How Low Should You Go?
The evidence for aggressive LDL targets: regression trials, physiological baselines, and why "lower is better" isn't just a slogan
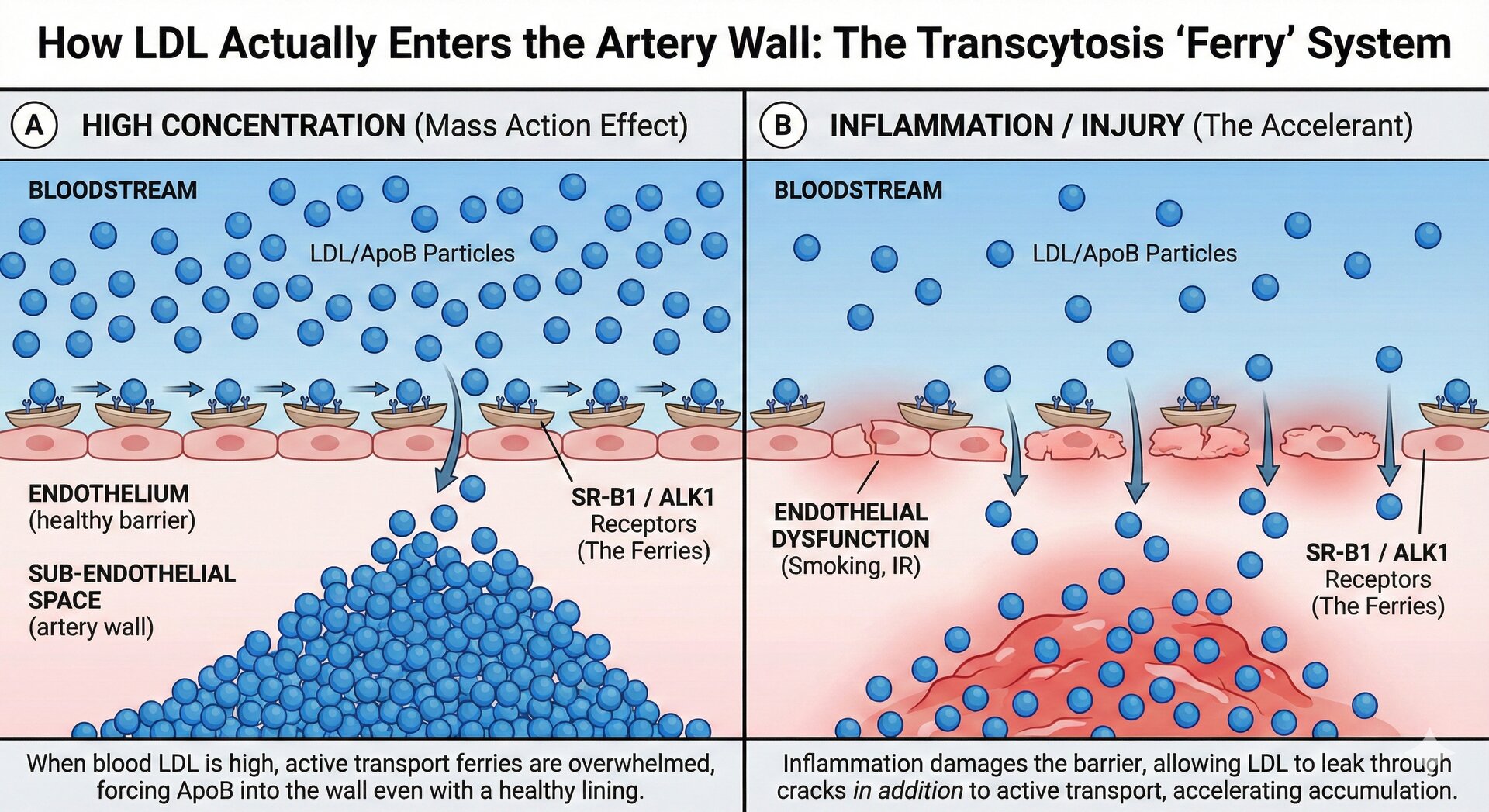
"At ~1.8 mmol/L LDL-C, plaques stop growing. Below that, they shrink. No floor has been found—0.5 mmol/L is better than 1.8."
TL;DR: The 60-Second Summary
- 1 Plaque regression begins below ~1.8 mmol/L LDL-C. Above that threshold, plaques grow. Below it, they shrink. This is proven by intravascular ultrasound studies (GLAGOV).
- 2 There's no J-curve or "too low." Patients at 0.5 mmol/L have better outcomes than those at 1.8. No cognitive or safety issues have emerged at ultra-low levels.
- 3 The physiological baseline is 0.8-1.8 mmol/L. Neonates have LDL of 0.8-1.3 mmol/L. Hunter-gatherers (Tsimane, Hadza) have 1.6-2.3 mmol/L. Our "normal" ranges are biologically abnormal.
- 4 Cumulative exposure matters—lower for longer wins. A genetic mutation giving lifetime LDL of 1.0 mmol/L prevents 88% of heart disease. Starting statins at 50 provides less benefit than starting at 30.
- 5 Current guidelines reflect this evidence. Very high risk: <1.4 mmol/L (ESC) or <1.8 mmol/L (ACC/AHA). Secondary prevention with recurrent events: <1.0 mmol/L may be optimal.
"Your LDL of 130 is fine—that's normal." Is it? The evidence says otherwise. Let's examine what the science actually shows about optimal LDL levels and why the targets have been getting lower over time.
The Regression Threshold
The GLAGOV Curve: Where Plaques Stop Growing
Relationship between achieved LDL-C and change in plaque volume (Percent Atheroma Volume)
mmol/L: Plaque GROWS
mmol/L: STABILIZATION
mmol/L: Plaque SHRINKS
The Takeaway
The ~1.8 mmol/L threshold isn't arbitrary—it's the point where the rate of LDL particle entry into the artery wall equals the rate of removal by reverse cholesterol transport. Below this, the artery can heal itself.
The Physiological Baseline
What's "normal" LDL? The answer depends on whether you mean statistically normal (average for modern Western populations) or biologically normal (what human physiology actually requires).
LDL-C Levels Across Populations
The Tsimane (Bolivia)
- Mean LDL-C: 1.8-2.3 mmol/L
- Coronary disease: Lowest ever recorded
- Inflammation: High (chronic infections)
- Implication: Low LDL protects even with inflammation
Neonates (Cord Blood)
- Mean LDL-C: 0.8-1.3 mmol/L
- Significance: Rapid growth phase
- Proof: Cells can build membranes at these levels
- Implication: Higher levels aren't "needed"
Cumulative Exposure: Area Under the Curve
Atherosclerosis is a cumulative disease. It's not just your LDL level today—it's the total exposure over decades. Think of it like sun damage: a lifetime of moderate sun is worse than a few intense days.
Lifetime LDL Exposure: Why Earlier is Better
PCSK9 Loss-of-Function Mutation
Lifetime LDL ~1.0 mmol/L → 88% risk reduction
Statin Started at Age 50
LDL to 1.8 mmol/L → 25-35% risk reduction
Familial Hypercholesterolemia: Proof of Dose-Response
People with FH have lifelong LDL of 5.2-13.0+ mmol/L. Untreated, they have heart attacks in their 30s-40s. This "natural experiment" proves that cumulative LDL exposure directly causes heart disease—and that genetics don't protect you from physics.
Current Guideline Targets
| Risk Category | ESC 2021 | ACC/AHA 2018 | Notes |
|---|---|---|---|
| Low Risk | <3.0 mmol/L | — | General population, no specific target |
| Moderate Risk | <2.6 mmol/L | 30-50% reduction | Some risk factors, no established disease |
| High Risk | <1.8 mmol/L | <1.8 mmol/L | Significant risk factors, diabetes, or CKD |
| Very High Risk | <1.4 mmol/L | <1.8 mmol/L | Established ASCVD, recurrent events |
| Extreme Risk | <1.0 mmol/L | — | Recurrent events on therapy (ESC consideration) |
Note on ApoB targets: ESC guidelines also specify ApoB targets: <0.65 g/L for very high risk, <0.8 g/L for high risk. ApoB is increasingly recognized as the better marker for treatment goals.
Addressing Common Concerns
"Won't very low LDL affect my brain?"
No. The brain makes its own cholesterol behind the blood-brain barrier—it doesn't import LDL from blood. FOURIER and other trials with patients at LDL levels of 0.5-0.8 mmol/L showed no cognitive effects. Neonates develop their brains normally at LDL 0.8-1.3 mmol/L.
"Isn't there a J-curve at very low levels?"
Observed J-curves in epidemiological studies reflect reverse causation: sick people (cancer, liver disease) have low cholesterol because of their illness, not the other way around. In randomized trials, where we lower cholesterol to these levels, there's no J-curve—only continued benefit.
"What about hormone production?"
Steroid hormones require cholesterol, but the amount needed is tiny compared to membrane synthesis. Cells can also synthesize their own cholesterol. At LDL levels of 0.4-0.8 mmol/L, testosterone and cortisol levels remain normal.
"If my CAC is zero, do I need a low LDL?"
CAC of zero means no calcified plaque—a late-stage finding. You may have early soft plaque or simply haven't accumulated enough "plaque-years" yet. High LDL with CAC=0 predicts future CAC development. The question is: do you want to intervene early or wait for damage?
Deep Dive: The Evidence Base
The following section provides comprehensive scientific detail for those who want to understand the primary evidence behind LDL targets.
The GLAGOV Trial: Proving Regression
The GLAGOV (GLobal Assessment of Plaque ReGression with a PCSK9 AntibOdy as Measured by IntraVascular Ultrasound) trial was a pivotal study that used intravascular ultrasound (IVUS) to measure the precise volume of atherosclerotic plaque in coronary arteries before and after intensive lipid-lowering therapy.
Patients were randomized to receive evolocumab (a PCSK9 inhibitor) or placebo on top of statin therapy. The primary endpoint was change in Percent Atheroma Volume (PAV)—the percentage of the vessel wall occupied by plaque.
Key Findings
The study demonstrated a strictly linear relationship between achieved LDL-C levels and plaque volume change:
- The Zero-Crossing Point: The regression line crosses zero (no progression) at approximately 1.8 mmol/L. Above this level, plaques grow; below it, they regress.
- No Floor Effect: Patients achieving LDL-C as low as 0.5 mmol/L achieved significantly greater plaque regression than those at 1.8 mmol/L. The relationship remained linear throughout the range studied.
- Regression Rate: For every 0.3 mmol/L reduction in LDL-C below 1.8 mmol/L, there was additional plaque regression.
- Safety: No significant safety signals were observed at ultra-low LDL levels, including no cognitive effects or increases in hemorrhagic stroke.
This data provides the mechanistic basis for aggressive LDL targets: there exists a biological threshold below which the artery can heal. The exact threshold (~1.8 mmol/L) represents the point where LDL particle entry rate equals the rate of removal via reverse cholesterol transport.
Mendelian Randomization: Nature's Trials
Mendelian randomization uses genetic variants as "natural experiments" to determine causality. Because genetic variants are assigned randomly at conception, they're not subject to confounding the way observational studies are.
PCSK9 Loss-of-Function Mutations
Approximately 2-3% of African Americans carry loss-of-function mutations in PCSK9 that result in lifelong LDL-C levels approximately 0.8-1.0 mmol/L lower than non-carriers. The Dallas Heart Study and subsequent analyses showed:
- 28% lower LDL-C from birth
- 88% reduction in coronary heart disease risk
- No adverse health effects
This disproportionate benefit (88% risk reduction from 28% LDL reduction) illustrates the power of cumulative exposure. The same LDL reduction achieved with a statin at age 50 provides only 25-30% risk reduction—because decades of exposure have already occurred.
HMGCR Variants and Statin-Like Effects
Genetic variants in HMGCR (the target of statins) that lower LDL-C by ~0.3 mmol/L are associated with proportional reductions in cardiovascular risk—supporting the mechanism of statin benefit and the causal role of LDL-C.
The Hunter-Gatherer Evidence
Populations living in environments approximating the human evolutionary niche provide insight into physiological lipid levels free from modern dietary influences.
The Tsimane
The Tsimane people of the Bolivian Amazon represent a unique natural experiment. Despite living conditions that drive chronic inflammation (evidenced by elevated hs-CRP levels, often exceeding 3 mg/L due to high infectious burden), they have the lowest prevalence of coronary artery atherosclerosis ever recorded.
- Mean LDL-C: 1.8-2.3 mmol/L
- CAC scores: Near-zero prevalence of significant coronary calcium
- Implication: This population proves that low LDL provides protection even in the presence of high inflammation—supporting the primacy of the lipid substrate over inflammatory triggers.
The Hadza
The Hadza hunter-gatherers of Tanzania maintain mean LDL-C levels around 1.6 mmol/L, with similarly low rates of cardiovascular disease.
Neonatal Data
Human neonates, who are undergoing the most rapid phase of growth in the human lifecycle (including brain development), have cord blood LDL-C levels of 0.8-1.3 mmol/L. This confirms that all physiological functions—cell membrane synthesis, steroid hormone production, bile acid formation—can be met at these concentrations.
Familial Hypercholesterolemia: The Dose-Response Proof
If cumulative LDL exposure causes atherosclerosis, then lifelong extreme elevations should cause early disease. Familial hypercholesterolemia (FH) confirms this prediction:
- Heterozygous FH: Lifelong LDL 5.2-9.0 mmol/L → Untreated, 50% of men have heart attacks by age 50
- Homozygous FH: Lifelong LDL 13-26+ mmol/L → Untreated, heart attacks occur in childhood/teens
FH provides a natural dose-response curve proving that higher LDL = earlier disease. The only variable is LDL level—these individuals don't have more inflammation or other risk factors. Their LDL is simply higher from birth.
The FOURIER Trial: Ultra-Low Safety
FOURIER randomized 27,564 patients with atherosclerotic cardiovascular disease to evolocumab (a PCSK9 inhibitor) or placebo on background statin therapy. The evolocumab group achieved median LDL-C of 0.8 mmol/L, with many patients reaching levels of 0.4-0.6 mmol/L.
Safety Findings at Ultra-Low Levels
- No increased hemorrhagic stroke: A theoretical concern based on observational data was not observed
- No cognitive effects: Extensive neurocognitive testing showed no differences from placebo
- No diabetes increase: Unlike statins, PCSK9 inhibitors don't increase diabetes risk
- No cancer increase: Follow-up showed no signal for malignancy
The FOURIER trial provided crucial evidence that achieving LDL levels well below current targets is safe and provides additional benefit.
The "Lower is Better" Controversy: Addressing Counterarguments
The Observational J-Curve
Critics point to observational studies showing increased mortality at very low cholesterol levels. However, this relationship reflects reverse causation: serious illnesses (cancer, liver failure, malnutrition, chronic infections) lower cholesterol levels. When we randomize patients to achieve low levels through medication, no J-curve appears.
Lean Mass Hyper-Responders (LMHRs)
The KETO-CTA study examined individuals on ketogenic diets with extreme LDL elevations (>5.2 mmol/L) but pristine metabolic profiles (high HDL, low triglycerides, low inflammation). Short-term data showed less plaque progression than expected. However:
- Mean diet duration was only 4.7 years—insufficient to detect slow accumulation
- Patients with baseline plaque still progressed
- This doesn't negate the causal role of LDL; it suggests metabolic health may raise the threshold for harm, but not eliminate it
The CAC = 0 Paradox
Some argue that high LDL with CAC = 0 means the LDL isn't harmful. However:
- CAC measures calcified plaque—a late finding. Soft plaque may be present but unmeasured.
- High LDL with CAC = 0 strongly predicts future CAC development
- The question is whether to wait for damage or prevent it
Synthesis: The Evidence-Based Position
The convergence of evidence from IVUS regression trials, Mendelian randomization, population studies, and clinical outcomes trials supports a clear conclusion:
- Approximately 1.8 mmol/L is the threshold where plaques stop growing
- Lower is consistently better down to at least 0.5 mmol/L, with no floor effect observed
- 0.8-1.8 mmol/L represents the physiological range humans evolved with
- Cumulative exposure (area under the curve) determines lifetime risk
- Current "normal" ranges (2.6-3.4 mmol/L) are statistically average but biologically abnormal
For individuals with established cardiovascular disease or high-risk features, targets of <1.4 mmol/L (ESC) or <1.8 mmol/L (ACC/AHA) are evidence-based. For those with recurrent events despite therapy, targeting <1.0 mmol/L may be optimal.
References
- Nicholls SJ, Puri R, Anderson T, et al. Effect of Evolocumab on Progression of Coronary Disease in Statin-Treated Patients: The GLAGOV Randomized Clinical Trial. JAMA. 2016;316(22):2373-2384. PubMed →
- Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264-1272. PubMed →
- Ference BA, Yoo W, Alesh I, et al. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a Mendelian randomization analysis. J Am Coll Cardiol. 2012;60(25):2631-2639. PubMed →
- Kaplan H, Thompson RC, Trumble BC, et al. Coronary atherosclerosis in indigenous South American Tsimane: a cross-sectional cohort study. Lancet. 2017;389(10080):1730-1739. PubMed →
- Pontzer H, Wood BM, Raichlen DA. Hunter-gatherers as models in public health. Obes Rev. 2018;19 Suppl 1:24-35. PubMed →
- Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med. 2017;376(18):1713-1722. PubMed →
- Giugliano RP, Mach F, Zavitz K, et al. Cognitive Function in a Randomized Trial of Evolocumab. N Engl J Med. 2017;377(7):633-643. PubMed →
- Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41(1):111-188. PubMed →
- Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol. J Am Coll Cardiol. 2019;73(24):e285-e350. PubMed →
- Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. Eur Heart J. 2017;38(32):2459-2472. PubMed →
- Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease. Eur Heart J. 2013;34(45):3478-3490a. PubMed →
- Nissen SE, Nicholls SJ, Sipahi I, et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA. 2006;295(13):1556-1565. PubMed →
- Blom DJ, Hala T, Bolber M, et al. A 52-week placebo-controlled trial of evolocumab in hyperlipidemia. N Engl J Med. 2014;370(19):1809-1819. PubMed →
- Norwitz NG, Soto-Mota A, Kaber B, et al. The Lipid Energy Model: Reimagining Lipoprotein Function in the Context of Carbohydrate-Restricted Diets. Metabolites. 2022;12(5):460. PubMed →
- Mitchell PD, Haw TJ, Rimm EB, et al. Zero coronary artery calcium testing and clinical outcomes. JACC Cardiovasc Imaging. 2019;12(8):1640-1642. PubMed →